L'emocromatosi giovanile si sdoppia
L'emocromatosi giovanile è una rara e grave forma di emocromatosi ereditaria distinta dall'emocromatosi classica HFE. Si manifesta precocemente, prima dei 30 anni, spesso già attorno ai 20 anni. Proprio per la giovane età di insorgenza del quadro clinico, la malattia si differenzia dall'emocromatosi classica dove i sintomi compaiono in genere dopo i 40-50 anni. Il sovraccarico di ferro è assai rilevante e precoce e coinvolge tutto l'organismo ma in particolare cuore, fegato, pancreas e ipofisi e comporta manifestazioni cliniche severe quali insufficienza cardiaca, cirrosi epatica, diabete, ipogonadismo ed un elevato rischio di mortalità.
La diagnosi di emocromatosi giovanile si basa sempre in prima istanza sull'analisi degli indici del ferro, ma richiede comunque la conferma con la biopsia epatica. E' molto importante che la diagnosi sia precoce, perchè se si interviene tempestivamente attraverso un intenso regime di salassoterapia la prognosi è buona.
L'emocromatosi giovanile è una malattia ereditaria recessiva. Ciò significa che i genitori del soggetto affetto sono entrambi portatori della malattia ma non sviluppano i sintomi, mentre i figli che ereditano il difetto genetico da entrambi i genitori sono malati. Fino a poco tempo fa si pensava che il difetto genetico responsabile della malattia fosse a carico di un unico gene localizzato sul cromosoma: (numero 1). Recentemente è stata invece identificata un'altra forma di emocromatosi giovanile provocata da un difetto genetico a carico di un diverso cromosoma: (numero 19). La scoperta è frutto della collaborazione tra l'équipe di Torino, coordinata dalla Prof.ssa C. Camaschella e il Dipartimento di medicina dell'Università di Atene.
La scoperta è interessante perchè il gene incriminato serve per produrre una
piccola proteina chiamata epcidina.
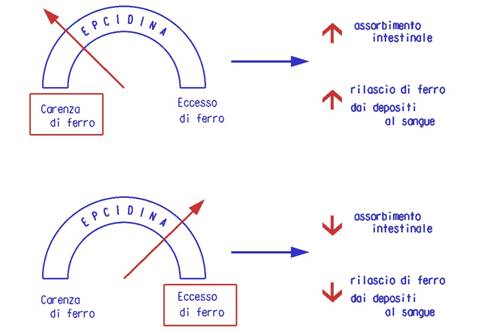
In condizioni normali l'epcidina agisce come un sensore che regola la
disponibilità del ferro in base alle necessità: nel caso di carenza di ferro
determinando un maggior assorbimento di ferro dalla dieta e un maggior rilascio
del ferro al sangue dai sistemi di deposito e, viceversa, in condizioni di
eccesso di ferro limitandone l'assorbimento o la sua dismissione in circolo dai
depositi. Quando il gene dell'epcidina è mutato questo sistema di regolazione
del ferro in relazione alle necessità viene meno: il risultato è un assorbimento
sregolato del ferro, un suo accumulo in diversi tessuti e il danno d'organo.
La mutazione fino ad oggi è stata identificata in una famiglia di origine greca
e in una di origine italiana ma andrebbe ricercata in tutti gli affetti dalla
forma giovanile di emocromatosi non legata al cromosoma 1.
L'emocromatosi giovanile, dato che non dipende dal gene HFE responsabile della
forma classica della malattia, viene oggi classificata all'interno del gruppo
delle emocromatosi non-HFE correlate. Nell'ambito di questo gruppo i casi
definiti geneticamente sono ancora purtroppo rari. La causa del sovraccarico
marziale nella maggior parte dei soggetti affetti da emocromatosi non-HFE è
infatti ancora non nota.
Risulta necessario pertanto lo sviluppo di sistemi rapidi ed efficaci volti allo
studio delle mutazioni esistenti e di sistemi atti all'identificazione di nuove
mutazioni.
Riferimenti bibliografici:
A. Roetto, G. Papanikolaou et al. Nature Genetics 2003.
N.C. Andrews. Seminars in Hematology 2002.
[Articolo pubblicato il 17-04-03]